
- 23 Mar 2026
- Sam Whitmarsh
From Experiment-Centric to Data-Centric Optimisation: Introducing CHETAH – The Digital Core of Smarter, Faster High-Throughput Experimentation

This paper is the first part of a new CatSci series exploring the science, strategy, and technology behind our approach to High-Throughput Experimentation (HTE). Each instalment will examine a different aspect of the workflow – from design to automation to analytics and the platform that holds all this together.
In this first paper, we outline CatSci’s approach to experimental design within its fully digital high-throughput experimentation (HTE) workflow. It focuses on how four complementary design paradigms – Screening, DoE, Bayesian optimisation, and AI-guided design – are integrated through a central orchestration platform that connects design, automation, and analytics. Together, these methods enable faster experimentation, richer data capture, and deeper scientific understanding of reaction variables and their interactions. By building a structured experimental database, CatSci is creating a foundation for intelligent seeding and pre-trained predictive models that will guide future development. This work represents part of our ongoing transformation in experimental chemistry from experiment-centric to data-centric optimisation, accelerating innovation across our development projects. To ensure scalability and consistency across our Chemistry and Analytical functions, we created the CHETAH Platform – Chemistry High-Throughput Testing, Experimentation, Automation, and Harmonisation – a digital orchestration environment that integrates design, automation, and analytics to accelerate decision-making and knowledge capture.
1. Introduction
Over the past two decades and more, automation and HTE has transformed the pace and scale of chemical development. Modern laboratories can now generate data at rates once unthinkable, integrating parallel synthesis, robotic solid and liquid handling, and rapid analytical workflows into unified, automated systems (Mennen et al, OPRD, 2019), (Lu et al, AIChem, 2024). These advances have enabled chemists to explore chemical space more quickly and more reproducibly than ever before. Yet across the industry, throughput alone is not enough. As Gaunt and co-workers note, “automation without design produces only data, not understanding” (Gaunt et al, Chem, 2021). The true differentiator lies not in how quickly experiments can be run, but in how efficiently value is derived from them – this in turn comes from how intelligently they are designed.
At CatSci, our HTE workflow connects each experimental component, from digital design through robotic weighing and liquid handling, to automated analytical sequence generation and processing, to reaction data visualisation, creating a fully integrated cloud based, design–make–test–analyse (DMTA) environment. Within this system, the design stage acts as the primary intelligence layer: the point where scientific intent is translated into structured, executable plans that drive automation downstream.
This paper focuses on that design component. It explores how four complementary approaches: Screening, Design of Experiments (DoE), Bayesian optimisation, and AI-guided design, form a suite of approaches that support our team to learn more quickly, explore more broadly, and optimise more efficiently. We’ll discuss why each component has been chosen and the strengths and drawbacks of each.
2. The evolution of experimental design in chemistry
Experimental design in chemistry has developed through several distinct paradigms, each offering a different way to balance creativity, efficiency, and statistical rigour. Rather than displacing one another, these paradigms coexist, each suited to different problems and stages of chemical discovery and development. We’ve used a review paper by Taylor et al. (Chem. Rev., 2023), to group these approaches as they broaden from empirical and statistically structured designs to adaptive, data-driven approaches:
The first paradigm, grounded in empirical exploration, relies on chemical intuition and one-variable (or factor)-at-a-time (OVAT/OFAT) variation. These methods remain valuable for the rapid screening of a small chemical space or hypothesis generation when knowledge is limited, materials are scarce, or instrumentation is constrained. This approach is simple to understand, is very familiar from undergraduate studies and allows chemists to explore comfortably, though at the cost of efficiency and completeness – especially for large design spaces and relationship between variables.
The second paradigm, defined by statistically structured design, represents the enduring strength of Design of Experiments (DoE). Factorial, D-optimal and response-surface designs provide interpretability, reproducibility, and statistical confidence. They underpin the Quality-by-Design principles established by regulatory bodies and continue to form the backbone of robust process development across the pharmaceutical industry. This approach remains highly familiar to industrial chemists, offering a structured way to quantify how variables interact and influence outcomes. Manual DoE remains practical when exploring three to four variables, but beyond this, experimental complexity can turn expert scientists into sampling operators. Robotic automation extends that range, typically enabling the study of around three to eight variables in a single design, depending on whether full factorial, response-surface, or fractional strategies are used. A 5-variable study with 3 factors and some centre replicates and error estimation might equal 260 experiments (full factorial), that needs 3×96 well plates. With 4-5 solid components in a typical catalysis screen that’s ~ 1300 individual weighing operations – a sizable effort even for current automation. Even with more efficient design strategies in DoE, such as fraction of DSD, as the number of variables increases, the number of model terms grows rapidly, and maintaining orthogonality among factors becomes more difficult. The resulting matrix can become less well-conditioned, increasing variance in the estimated coefficients and reducing the precision and interpretability of the fitted model.
The third paradigm in the Taylor et al. (Chem. Rev., 2023) paper: Adaptive learning, algorithmic design and autonomous experimentation, extends these principles into dynamic learning. Bayesian optimisation, active learning and reinforcement strategies use probabilistic surrogate models that evolve with each experimental result and guide subsequent runs toward regions of highest information or improvement. This approach can efficiently manage 10+ variables, balancing exploration with exploitation and dramatically reducing experiment counts. However, it often sacrifices the transparency, orthogonality and straightforward variance analysis that make DoE so interpretable and regulator-friendly. Adaptive methods deliver speed and scalability, but DoE still provides the clearest window into causality.
In our automated high-throughput experimentation platform, we have designed the workflow so that scientists can easily access all three paradigms, particularly the second and third, by integrating Screening, DoE, Bayesian, and AI, guided approaches within a single digital environment. This flexibility ensures that the right design tool can be applied to the right problem at the right time.
The following section outlines each of these design modes in turn, explaining their purpose, strengths, and how they connect together within the experimental framework.
3. The four design modes (and one extra)
CatSci’s experimental design framework brings the 3 paradigms together in four complementary approaches within one unified digital workflow: Screening, Design of Experiments (DoE), Bayesian optimisation, and AI-guided design. Predicitive AI is not currently a mature deployable technology (at least in our hands).
3.1. Screening: User-defined exploration and statistical seeding
User defined screening enables chemists to rapidly explore a defined reaction or process space. The scientist specifies the factors, levels, and constraints of interest, and the platform produces a feasible design – generating plate layouts, weighing instructions, and analytical sequences automatically. This provides a simple, reproducible way to survey custom condition sets, ideal for early-stage route scouting or reactivity mapping.
This approach can be broadened to a seeding stage that acts as a precursor to further experimental designs. Here the user defines the edges of a design space which is then broken down algorithmically using a Sobol sequence to generate quasi-random points that uniformly cover the factor space without bias or redundancy. This creates an efficient, unweighted low resolution map of even very large experimental domains, ensuring that initial results capture the full diversity of behaviours. The resulting design is not particularly interpretable by a human, especially if there is high dimensionality as the patterns and trends are often obscured by the quasi-random structure. But it does provide a statistically rich and unbiased dataset that forms the foundation for subsequent second, and third, paradigm approaches such as DoE and Bayesian optimisation.
3.2. Design of Experiments: Structured understanding and reproducibility
Design of Experiments (DoE) represents the core of the design framework and remains the statistical backbone of process understanding. After screening or seeding identifies a viable region of chemistry, DoE provides a structured way to quantify how variables influence key responses such as yield, purity, or selectivity.
Different design types are deployed according to the scientific question. Screening designs, such as fractional factorial or definitive screening designs, are used when the aim is to identify the few critical variables that drive performance. Optimisation designs, including central composite and Box–Behnken designs, map the curvature of response surfaces and enable prediction of optimal conditions across multi-dimensional spaces. For late-stage development, robustness designs test sensitivity around target conditions, confirming process resilience and defining the design space for control strategy definition.
CatSci’s digital platform supports both continuous and categorical factors within the same framework. Continuous variables (e.g. temperature, concentration, stoichiometry) are explored across defined ranges, while categorical factors (e.g. catalyst, solvent, ligand) are encoded appropriately to preserve orthogonality and comparability. Automated liquid handling and solid dispensing execute these designs reproducibly, ensuring accurate dosing and complete traceability from design intent through to analytical result.
The power of DoE lies in its interpretability and regulatory alignment. Each coefficient, interaction, and curvature term can be traced back to experimental evidence, forming part of the scientific rationale behind process control and validation. Within the workflow, the complete DoE design, raw data, fitted model, and analytical reports are connected and can be shared. This ensures that results can be reproduced, reviewed, and integrated directly into the technical documentation that supports regulatory submissions and Quality-by-Design filings if needed.
3.3. Bayesian Optimisation: Adaptive learning and efficiency
Bayesian optimisation (BO) is one approach that can be taken from the tertiary paradigm in the workflow. Rather than running every possible combination of conditions, BO learns from the results of completed experiments to decide which experiments to run next. A statistical model predicts both the expected outcome and the level of uncertainty across the design space, allowing the system to focus on experiments that are most informative or most likely to improve performance. This can dramatically reduce the number of experiments needed to reach meaningful conclusions.
CatSci’s implementation builds on Merck’s BayBE platform, which can handle both continuous and categorical variables within the same optimisation loop. Continuous factors such as temperature or reagent ratio are treated numerically, while categorical choices – like catalyst, ligand or solvent – are converted into chemical fingerprints using tools such as Mordred, RDKit, or ECFP descriptors. These descriptors translate molecular structure into numerical form, allowing the optimiser to recognise similarities between options and make more chemically informed suggestions rather than treating each category as unrelated.
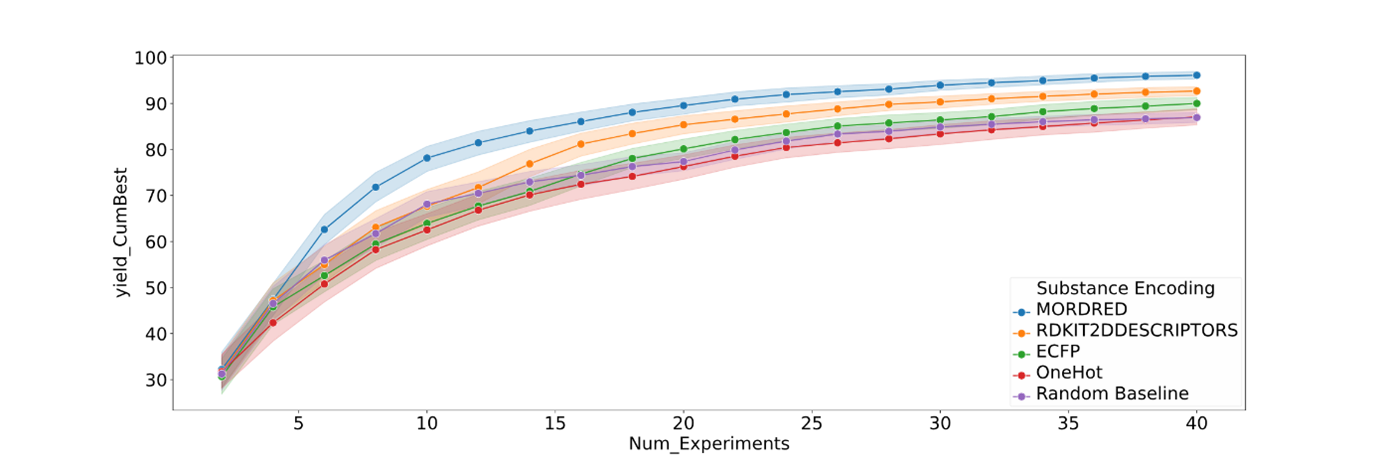
Figure 2 shows how different chemical encoding strategies influence optimisation efficiency when solvents, bases, or ligands are treated as categorical variables. Each curve represents the cumulative improvement in model performance as experiments progress. Descriptor-based encodings such as Mordred and RDKit2D achieve faster learning and higher final performance compared with one-hot (digitised columns for each categorical variable) or random baselines. This demonstrates that encoding chemical categories using structural and physicochemical features allows the optimiser to recognise similarities between molecules, generalise more effectively, and reach optimal regions of chemical space with fewer experiments.
The Bayesian loop begins with seed data (from Sobol, screening or DoE), retrains the model after each batch of experiments, and proposes new conditions that balance predicted improvement and uncertainty. For categorical factors, recommendations are made in the descriptor space but automatically mapped back to real chemical choices for execution. The system integrates visual analytics, uncertainty heat maps, predicted, versus, observed plots, and feature, importance summaries, so scientists can interpret which factors are most influential and where confidence is highest.
Like any iterative process, BO has trade, offs. It can be slower at the start while the model learns, and it is not always obvious when sufficient data have been gathered. This can be addressed with stopping rules based on model convergence and uncertainty thresholds, ensuring that campaigns conclude efficiently once the model stabilises and further experiments add little new information. We are still learning in this area and welcome insight from the community.
3.4. AI-Guided Design: Representation learning and coreset optimisation
The final stage of experimental design framework is AI-guided design, which builds on Bayesian optimisation but goes further by learning how the entire reaction space behaves, not just where the next best result might be. Our current deployment focuses on a method inspired by the RS-Coreset approach (Hua et al., Communications Chemistry, 2025), which uses representation learning and coreset selection to predict reaction outcomes from only a small fraction of possible experiments. A schematic is shown in figure 3.
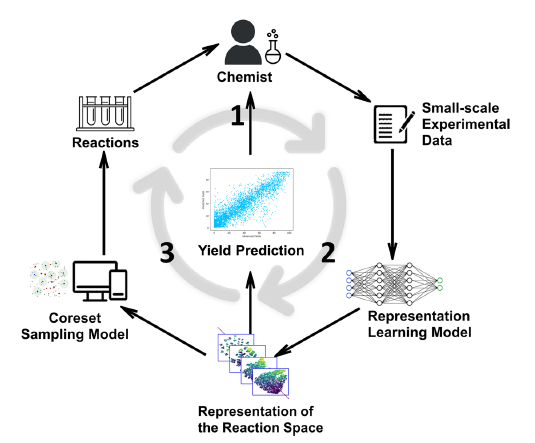
Figure 3 explains the experimental cycle. Step 1: Chemists evaluate reaction yields through small-scale experiments, generating new data. Step 2: A representation-learning model updates its understanding of the reaction space based on this experimental data. Step 3: A coreset sampling algorithm selects the most informative new reactions to test next. Repeating this loop refines the model’s internal representation of chemical space, enabling progressively more accurate yield prediction and efficient exploration of reaction conditions.
To explain simply, imagine mapping a new city:
- A full factorial DoE is like visiting every street – thorough but slow.
- Bayesian optimisation is like using a GPS that guides you toward promising neighbourhoods.
- The RS, Coreset method instead learns the city’s overall layout by sending scouts to just a few, diverse locations, then uses that information to predict what the rest looks like.
In practice, each reaction combination – defined by ligands, solvents, catalysts, bases, and substrates – is converted into a numerical fingerprint using molecular descriptor tools such as ECFP, Mordred, or RDKit – just like the Merck BO approach. These capture structural and physicochemical features so that both categorical and continuous variables can be represented in a shared numerical space. The model then selects a small, diverse subset of experiments (its coreset) to perform, learns from their outcomes, and refines its internal map of reactivity after each round.
Unlike DoE, which explicitly models each factor, or Bayesian optimisation, which focuses on incremental improvement, this AI, guided paradigm aims to capture the underlying landscape of chemical behaviour. It can recover high, performing conditions using as little as 2–5 % of all possible experiments.
The trade-off is that the model’s internal reasoning is less interpretable, it describes relationships in a high, dimensional latent space rather than with visible coefficients, but the gain in data efficiency and predictive power is significant. Within our third, paradigm workflow, this approach fills the exploratory niche, allowing AI to extend learning beyond tested data and transform early experimental results into generalisable chemical insight.
3.5. Predictive AI: Modelling molecular space for reaction yield prediction (not working for us yet)
An additional branch of the third paradigm design framework (or you might even call this the fourth paradigm) explores predictive AI, where the aim is to anticipate experimental outcomes before entering the laboratory. In this approach, machine-learning models are trained on large literature datasets to predict reaction yield directly from molecular structure and reaction conditions. The goal is not to replace experimentation, but to filter chemical space – directing human and robotic effort toward regions with higher probability of success and away from those with lower probability of delivering valuable results.
Our work has focused on well-studied transformations such as Suzuki–Miyaura and Buchwald–Hartwig couplings, using open-source frameworks including models described by Ahneman et al. (IOP Sci, 2020) and Schwaller et al. (J. Cheminf., 2023). The former applies random-forest regression on molecular descriptors and reaction conditions to predict yield across diverse cross, couplings, while the latter uses graph neural networks and transformer architectures to learn reactivity patterns directly from SMILES strings and contextual reaction metadata. Both represent state-of-the-art attempts to translate chemical structure into quantitative yield predictions.
In our internal benchmarking, however, neither descriptor-based nor neural models transferred reliably to our own laboratory data. When applied prospectively, predicted yields often diverged substantially from measured values, reflecting known challenges in generalising literature-derived models to industrial systems. As a result, we have paused deployment of ab initio predictive models and redirected our focus toward empirically guided optimisation using DoE, Bayesian, and AI-adaptive loops. Once larger, high-quality experimental datasets become available from our automation platforms, we plan to revisit predictive modelling – exploring pre-training and fine-tuning strategies that may finally bridge the gap between literature and laboratory performance.
4. Discussion: A Unified Framework for Intelligent Experimentation
Across this paper, we have described how CatSci’s high-throughput experimentation (HTE) platform unifies four complementary approaches to experimental design – Screening, DoE, Bayesian, and AI-guided experimentation. The fifth, exploratory stream in predictive AI is not yet mature. Each paradigm serves a distinct role in the design–make–test–analyse cycle. Screening offers seeding and rapid discovery, DoE delivers structured understanding, Bayesian optimisation delivers adaptive efficiency, and AI-guided design learns deeper patterns in extended data sets. If we can get Predictive AI to work(!), this will take this further, guiding where best to explore before experimentation even begins.
The strategic challenge is not to choose between the different paradigms, but to connect them effectively – they all have value when solving the right problem. In CatSci’s workflow, all design layers are linked through a central orchestration and automation hub that builds experimental plates, synchronises metadata, and integrates with weighing, liquid, handling, and analytical platforms. This ensures every stage, from screening to DoE, Bayesian learning, and AI-guided modelling, flows seamlessly as a single, traceable process. Human expertise remains central: scientists define the design space, select the most appropriate tool for the question at hand and use intuitive visualisation tools to interpret data and iterate results rapidly – importantly deciding when and why to stop or change direction. This combination of automation, data integrity, and expert insight accelerates our development projects while maintaining full scientific transparency.
5. Conclusions: Future Directions in Data-Centric Optimisation
The evolution from DoE to Bayesian and AI-driven design marks a transition from experiment-centric optimisation to data-centric discovery. CatSci’s goal is to build a continuously learning system where every experiment enriches a shared foundation of chemical knowledge.
While ab initio prediction models are still developing, our integrated empirical approach already delivers measurable benefits: faster optimisation, higher reproducibility, and improved decision-making. The next phase will strengthen this ecosystem, linking experimental design, execution, and analysis into a fully connected digital workflow.
We recognise that this is a rapidly advancing field shared by many laboratories worldwide. We warmly welcome advice, collaboration, and shared learning from others developing similar systems. By combining collective experience with CatSci’s platform-driven approach, we aim to accelerate the adoption of intelligent experimentation that enhances both scientific quality and operational efficiency across the chemistry community.